|
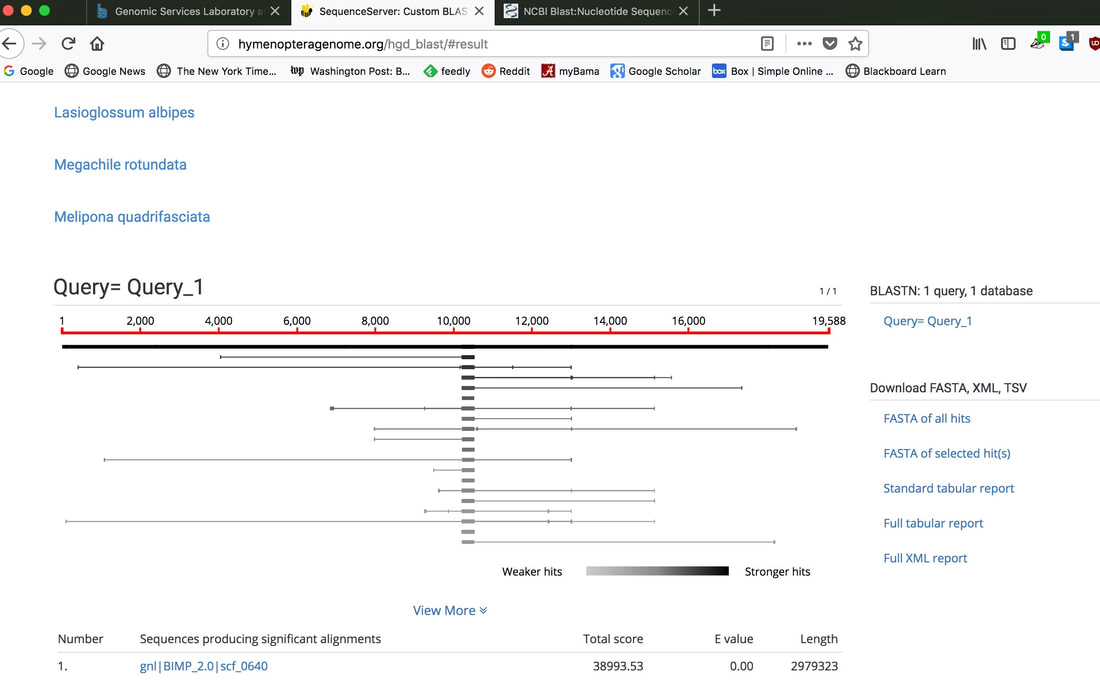
So continuing to play with our nanopore/illumina data in the search for a better reference genome for our target bumble bee taxa. Focusing on B. vosnesenskii here, I did a quick and dirty hybrid assembly that included ~30X Oxford Nanopore minion sequencing from 2 males from the same population, combined with ~100X HiSeq data from one of the same males. The assembly was done with the software MaSuRCA, which I gave ~300Gb RAM and 16 threads of computing power on the cluster, producing an assembly in roughly 3 days. So I'm playing with the data now (I'll probably add to this post as I get some other interesting results), but I just did some interesting exploratory analyses of synteny between my assembly and the two published bumble bees (B. impatiens, which is very scaffoldy, and B. terrestris, which has pretty good chromosome/linkage-group level assembly). I took one of my longer draft scaffolds for B. vosnesenskii and tried to see where it fit relative to other Bombus genomes using MAUVE. It appears that synteny is fully conserved for this ~4Mb chunk of genome (see top image), as expected for bumble bee genomes, but there are a few catches. First, this scaffold clearly belongs to the B. terrestris LG B10, which is ~13Mb in size. So clearly the minion+illumina data was't quite good enough to recover the whole chromosome. However, it does much better than the B. impatiens genome. In order to match up my scaffold, I had to stitch together 3 shorter B. impatiens scaffolds (see bottom image). This result looks like it might hold over the rest of the genome, given the distributions of scaffold lengths. So overall the hybrid assembly approach does a pretty darn good job, somewhere in between B. terrestris with its linkage group level assembly and B. impatiens with its 5000-odd scaffolds, without any mate-pair libraries etc. The grand total here = roughly $1,500 and a few days of work (plus the assistance of the Fierst and McKain labs, who got the instrument in the first place!). Of course, it will be much more work to annotate and stitch together the rest of the genome, but that's for a new grad student to work on! 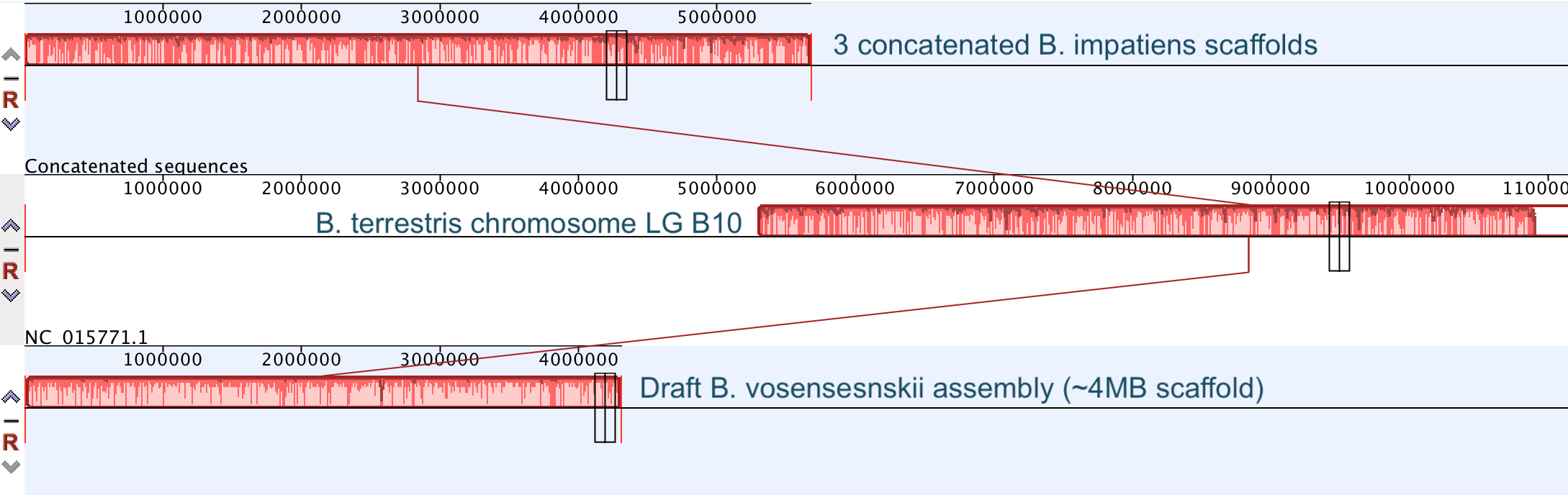 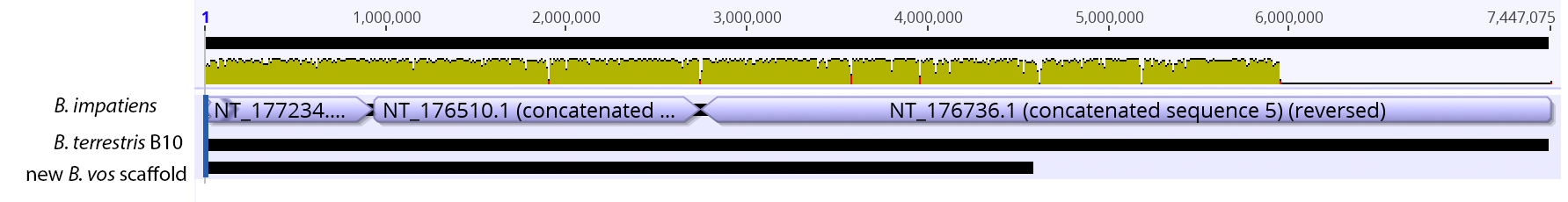 One additional thing we are seeing is that there can be a decent amount of contamination in the assembly, which is not necessarily surprising as we had to squish whole bees (including abdomens) to get enough DNA for the Nanopore runs. Numerous scaffolds stemm from bacteria, etc (most actually seem to be known bee symbionts or otherwise bee-associated, so these might be fun to look at downstream). We are playing around with some filtering applications but I think we've settled on the tool "BlobTools". This set of tools lets you map your raw reads (both Nanopore and Illumina) to the reference assembly, generate a BLAST database, and identify the origins of the contaminating sequences relatively quickly. You can then filter out anything you don't like using a combination of coverage, GC content, and taxonomy and redo your assembly. Just to visualize the effectiveness of cleaning the data you can compare a before and after plot (this is nanopore data). Pretty neat. One thing that's nice is that you can clearly see the big scaffolds in the center of the plot are nice and bee-y. Now to redo the assembly. 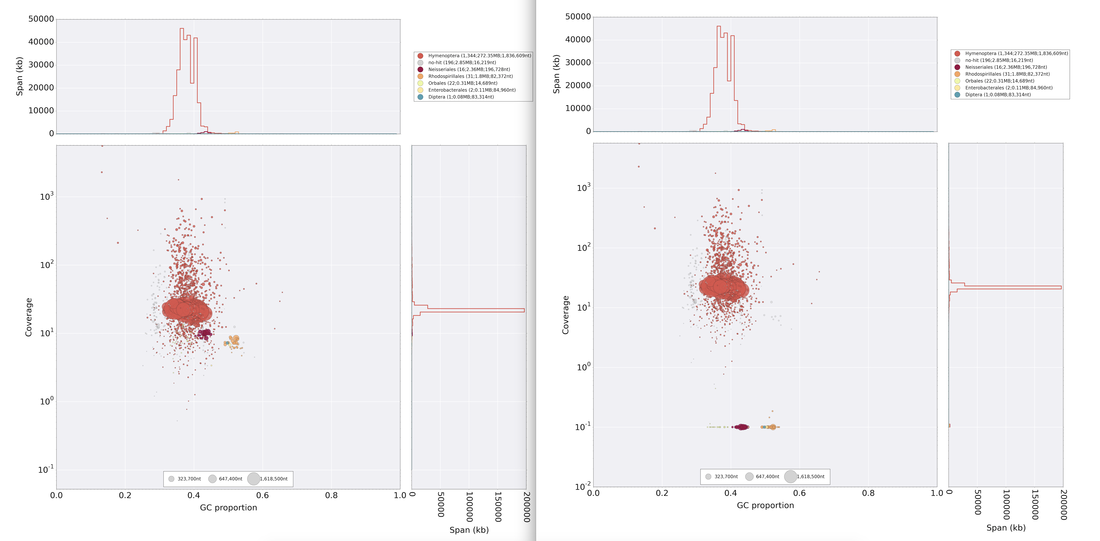 So in our effort to outdo ourselves in maxing out Oxford nanopore data we ran through a Bombus bifarius worker (black-banded form from the west coast) to get a genome assembly on which to align our low coverage Illumina WGS. We are quite happy with how this instrument is working for us for the long-read sequencing technology (haven't tried pac-bio yet, but we also don't have one of those upstairs) and are getting some nice data that should be sufficient for genome assemblies for B. vosnesenskii and B. bifarius. Just one run of B. vos (~10X coverage) seems to have given us an assembly with contigs with the integrity of the published B. impatiens genome, and we've gone ahead and done a second flowcell and Hiseq run for the same bee to clean things up. The new B. bifarius runs are really nice however, we got roughly 25Gb of sequence and nice long reads. The pic below is a single random read (yup that block of text is one read!) with a single BLAST hit all the way across against the B. impatiens genome with BeeBase...19kb!! Very exciting. We hope to get the B. vosensenskii and B. bifarius genomes put together over the next semester or so.  Thanks to John Sutton from the Fierst lab who is becoming an expert in all things nanopore and has been helping us out with the libraries and monitoring the instrument. Here's a pic of John and I very carefully doing Ampure bead cleanups of the libraries!
We previously attempted to sequence the B. vosnesenskii genome using Oxford Nanopore technology...didn't work out so good. Turns out we didn't know what we were doing! So we are trying again, thanks to Janna Fierst's intrepid grad student John Sutton and a few other students in the department who got some actual training on the thing. Things looking MUCH better so far, very good output, nice read length distribution etc! Turns out there are a few useful tricks for DNA isolation quality, etc. that aren't as important for sequencing with Illumina. Hopefully the data turn out to be useful for assembly, as we very much want to be able to align our population genetics data directly to a reference genome for our target species, as opposed to cross-species alignments against available genomes (e.g., Bombus impatiens) which works ok but has some challenges.  |
Lozier Lab NewsDispatches from the lab and field! Archives
March 2023
Categories
All
|

 RSS Feed
RSS Feed