|
Whole genome analyses reveal weak signatures of population structure and environmentally associated local adaptation in an important North American pollinator, the bumble bee Bombus vosnesenskii
Sam H Heraghty, Jeffrey D Lozier, Jason M Jackson bioRxiv 2023.03.06.531366; doi: https://doi.org/10.1101/2023.03.06.531366 A bit late but I'm currently trying to update the website and get everyone's papers updated! Sam recently published one of his dissertation chapters which is a cool look at whole genome variation linked to environmental variation across the B. vancouverensis range. Sam has a similar paper being submitted soon looking at a somewhat codistributed species Bombus vosnesenskii. 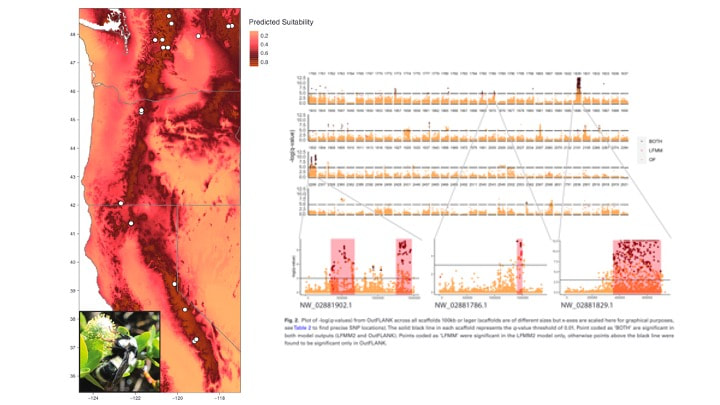 Sam D Heraghty, Sarthok Rasique Rahman, Jason M Jackson, Jeffrey D Lozier, Whole Genome Sequencing Reveals the Structure of Environment-Associated Divergence in a Broadly Distributed Montane Bumble Bee, Bombus vancouverensis, Insect Systematics and Diversity, Volume 6, Issue 5, September 2022, 5, https://doi.org/10.1093/isd/ixac025 We also published a followup study to look at how historical climate and range fluctuations shaped fluctuations in effective population size in B. vancouverensis to shape latitudinal gradients in diversity in this species. This is a cool example of linking ecological niche models to historical inferences from genome data using coalescent models to look at range-population size dynamics over time. 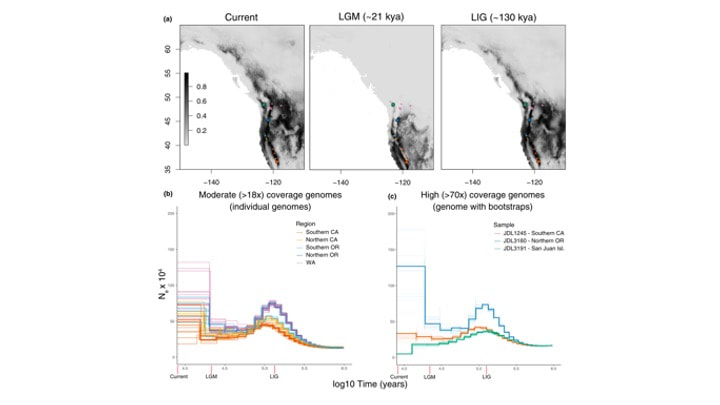 Lozier, J. D., Strange, J. P., & Heraghty, S. D. (2023). Whole genome demographic models indicate divergent effective population size histories shape contemporary genetic diversity gradients in a montane bumble bee. Ecology and Evolution, 13, e9778. https://doi.org/10.1002/ece3.9778
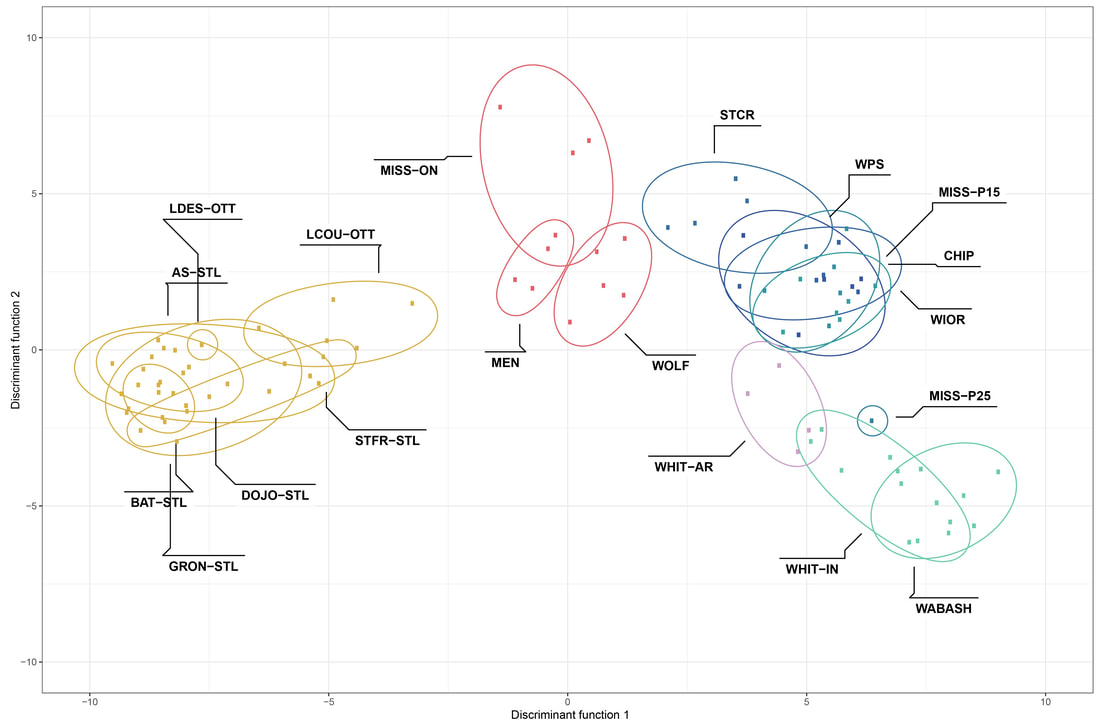
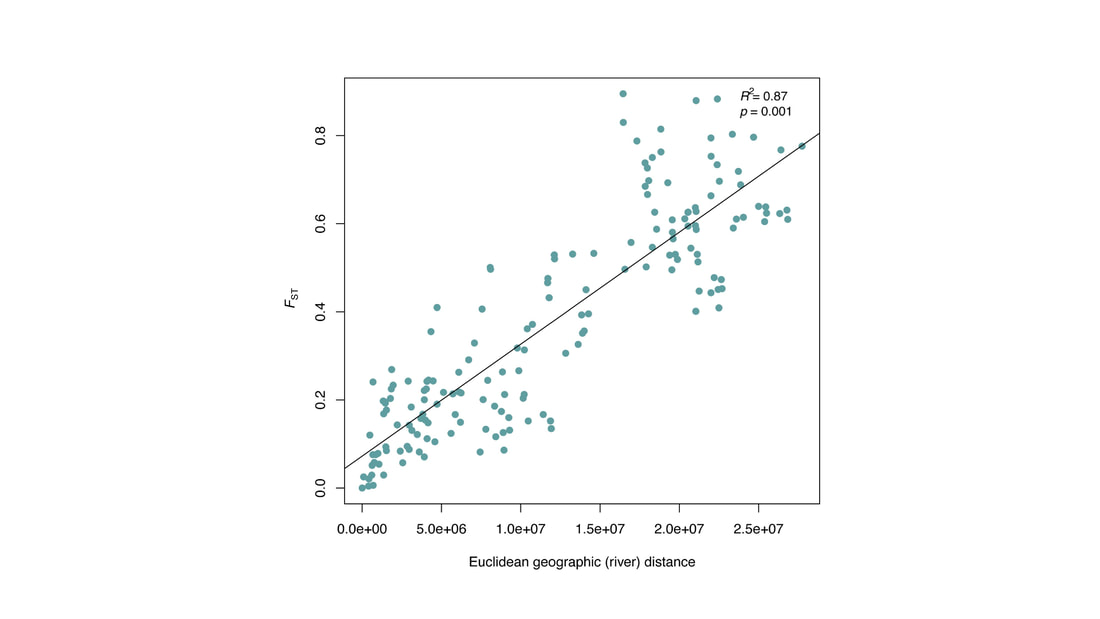
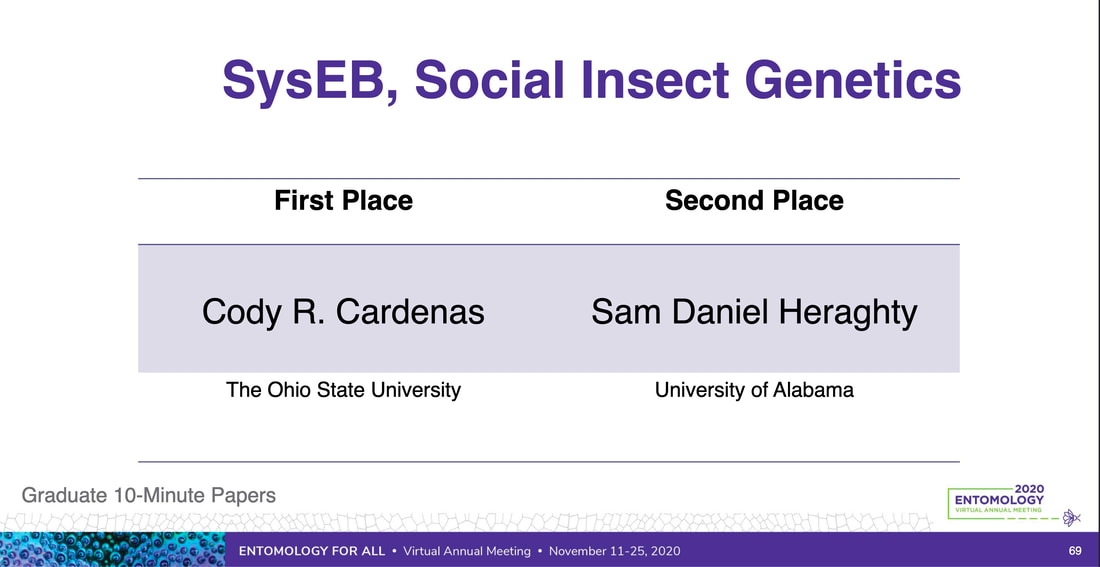
We just published a paper integrating population genomics (RADtag data generated by PhD student Jamie Bucholz) with microbiome data (from the Ole Miss DoB team) from Corbicula fluminea populations across the Mobile and TN river basins. This critter has some intriguing mechanisms of inheritance creating some unusual population genetic data, but overall we were able to pull out some interesting genetic patterns of structure in the species among rivers. The microbiome however, was unrelated to genetic structure of the clams, but was also structured by space and environment, and there was also seemingly an effect of the native mussel population on the C. flumina microbiome. The data suggest the microbiome may assist Corbicula in resource acquisition and facilitate invasion, but the processes structuring microbes and genetics of the populations differ during the invasion process. Chiarello, M., Bucholz, J. R., McCauley, M., Vaughn, S. N., Hopper, G. W., Sánchez González, I., Atkinson, C.L., Lozier, J. D., Jackson, C. R. (2022). Environment and Co-occurring Native Mussel Species, but Not Host Genetics, Impact the Microbiome of a Freshwater Invasive Species (Corbicula fluminea) . Frontiers in Microbiology. https://www.frontiersin.org/article/10.3389/fmicb.2022.800061 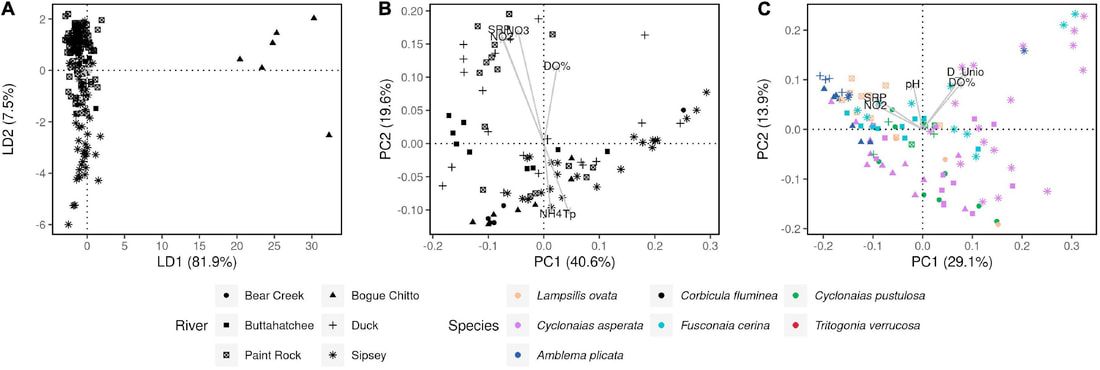 Congrats to Jamie Bucholz for acceptance of her RADseq study on Hickorynut mussels in Ecology and Evolution. Really cool results showing how genetic variation and structure varies with river distance and geographic barriers at large spatial scales in a species of conservation concern. Graduate students Sam Heraghty and Kelton Verble and postdoc Sarthok Rahman presented their research at the Entomology meeting last week in Denver and things seemed well received. In fact Kelton placed 2nd place in the president's prize competition for his talk, so congrats Kelton! We are looking for a PhD student (starting any time from Spring-Fall 2022) interested in large scale population-level genome sequencing of many US bumble bees. This is a collaboration with Heather Hines and Jonathan Koch, and the primary focus will be on species with color pattern variation, but there will be opportunities to analyze population genomics of bumble bees more generally. If sequencing lots of genomes from lots of bees from lots of species sounds like fun, shoot me an email! The full ad is pasted below. PhD Position in Comparative Population Genomics of Bumble Bees and their
Color Patterns: A PhD position is available in the laboratory of Jeff Lozier at The University of Alabama (lozierlab.ua.edu) as part of a recently awarded NSF project: “How many routes to the same phenotype? Genetic changes underlying parallel acquisition of mimetic color patterns across bumble bees”. This project is a collaboration with Dr Heather Hines at Penn State (hineslab.org) and Dr Jonathan Koch at the USDA Bee Lab in Logan, UT (jonathanbkoch.weebly.com). The PhD student will be involved in an interdisciplinary project to study the origins of color pattern variation in bumble bees. The focus of this position will be on comparative population genomics from whole genome resequencing of many North American bumble bee species. Range-wide whole genome data is already available in the lab for many bumble bee species, and the PhD student will be involved in additional field work and generation/analysis of high-throughput sequencing data. The student will also be able to make use of these extensive data sets to develop projects relating to conservation, evolutionary, and landscape genomics. We are looking to recruit a highly motivated student with interests in applying modern molecular and computational tools in a charismatic and ecologically important non-model group. Students will join an active, diverse, collaborative, and friendly lab (lozierlab.ua.edu/people.html) and department (U Alabama Biological Sciences: bsc.ua.edu). We also expect substantial opportunities for collaboration with all PIs and students/postdocs associated with the project. Contact Jeff Lozier ([email protected]) for more information. The position is available starting Spring 2022 but students interested in Summer or Fall 2022 are also encouraged to apply.  This week I took (yet another) road trip up to University of Illinois Urbana-Champaign to visit my post-doc advisor Sydney Cameron. We collected a lot of bees as part of our USDA Bumble bee decline project and we wanted to make sure the collection stayed all together in one freezer for the foreseeable future. So I packed up all the thousands of frozen bee samples representing many US Bombus in coolers of dry ice and lugged them back to UA. We hope to use these samples for several forthcoming projects relating to bumble bee evolution and conservation genomics.  I was also able to visit my friend Alex Harmon-Threatt who is a good buddy from back in Berkeley and is a prof in the Entomology Dept at UI that does cool stuff with pollinator biology.
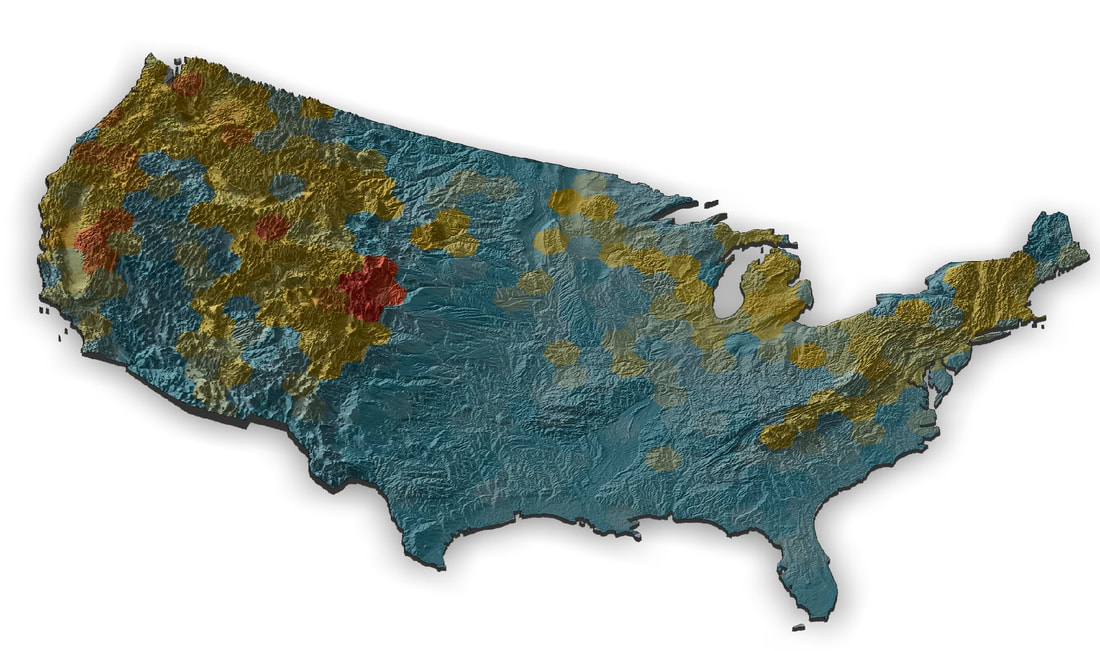 I'm working on a museum display as part of the outreach activities for our NSF Understanding the Rules of Life mountain epibeenomics grant and decided to illustrate why mountains are especially interesting for bumbles by making a map overlaying species richness with elevation. I just used some fairly heavily post-filtered data downloaded from GBIF for this, so could probably be easily improved on with some more rigorous filtering (although the richness blobs correlate pretty well with Paul Williams' maps, so they are pretty good I think). I determined a gridded richness estimate using R with mostly the sf and raster packages and made the pretty map with rayshader. It was a lot of work to wrangle the data and get all the layers in the correct formats, but came out pretty well I think.
PhD student Jamie Bucholz's poster took first place at the 2021 Freshwater Mollusk Conservation Society annual meeting!! Woot. Check it out over on mussels page. Update, Jamie got a nice plaque.   Say hello to Sarthok Rahman, the new postdoc that will work with Lozier and collaborator Janna Fierst on our Understanding the Rules of Life NSF grant on the epigenetics of cold tolerance across elevations. Sarthok comes to UA from Heather Hines' lab at Penn State, where he did some really great work on color pattern evolution in bumble bees. Sarthok is a bioinformatics whiz, and we hope he will make some major contributions to linking different levels of biological organization, from physiology, RNAseq, and methylation sequencing to understand the role of molecules in this phenotype!
He's already hard at work optimizing pipelines for bioinformatic analysis of bisulfite data, so we are excited to see what emerges from the data. Congratulations to UA BSC graduate Peter Scott, who went on to do great things as a postdoc out at UCLA, and is now an Assistant Professor at Assistant Professor at West Texas A&M Univ.
Peter just had a first-authored paper in Science (plus the cover!!). Peter studied tortoises that were part of a big relocation project out in southern CA and looked to try to predict what predicted survival of the relocated individuals. The idea is really simple, but provides excellent data that supports a classic assumption that underlies much of modern population/conservation genetics: more genetic variation = greater fitness. This is often difficult to test, but in this case they had the data on individual survival. They use extensive RAD-tag SNP set and look at probability of survival based on individual heterozygosity, translocation distance, and geographic origin... lo and behold, heterozygosity was the key predictor of tortoise survival. Really cool stuff. Congrats Peter! Scott, P. A., Allison, L. J., Field, K. J., Averill-Murray, R. C., & Shaffer, H. B. (2020). Individual heterozygosity predicts translocation success in threatened desert tortoises. Science, 370(6520), 1086–1089. https://doi.org/10.1126/science.abb0421 EntSoc was a little weird this year being all virtual and all, but it seems like the recorded talks were still well attended and the meeting was successful overall (I didn't attend this year because I'm pulling my hair out with regular academic duties...). That also didn't stop Sam Heraghty from giving a great talk on his population genomics work with bumbles. As part of the Mountain Bees Genome Project, Sam has sequenced hundreds of whole genomes from across the ranges of B. vosnesenskii and B. vancouverensis/B. bifarius to look for candidate genes adapted to climate using a landscape genomics approach (to expand on our prior work by Dr Jackson), and it was clearly well-received because he won second place for grad student talks! Papers coming soon! As part of our bee epigenetics NSF project we sequenced some bumble bee genomes, which included Oxford Nanopore Tech. long read sequencing data. We are playing around still, but tried out the pipeline using Nanopolish (available here) to directly identify CpG motifs and identify the fraction of called bases at each motif that appear to be chemically modified. So far I've tried it with the two B. vosnesenskii males (from colonies sourced from same site in OR) we used for the genomes, and see highly consistent results between the individuals across all the scaffods I've peeked at. We have some unpublished bisulfite data for workers from this species, as well as ONT data for other Pyrobombus so will be interesting to compare those data as well. 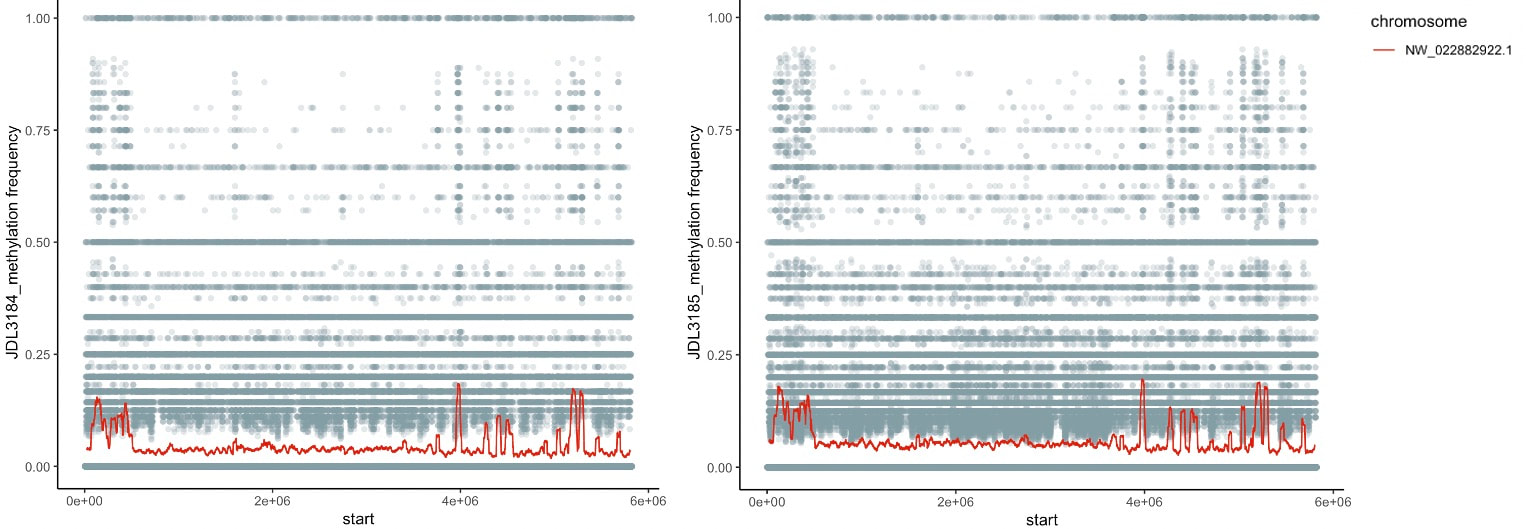 We finally published data from a longstanding project looking at cold adaptation in bumble bees. We started colonies from across the geographic range of B. vosnesenskii (across latitude and altitude) and measured thermal limits (CTmin and CTmax) as well as gene expression in all bees. We found a striking correlation of CTmin with annual mean temperature experienced by each population, and likewise found interesting correlations in gene expression in response to cold across the species range. Intriguingly there was little variation in CTmax or in the genomic response to warming, suggesting species may be constrained in terms of adaptability to increasing environmental temperatures. 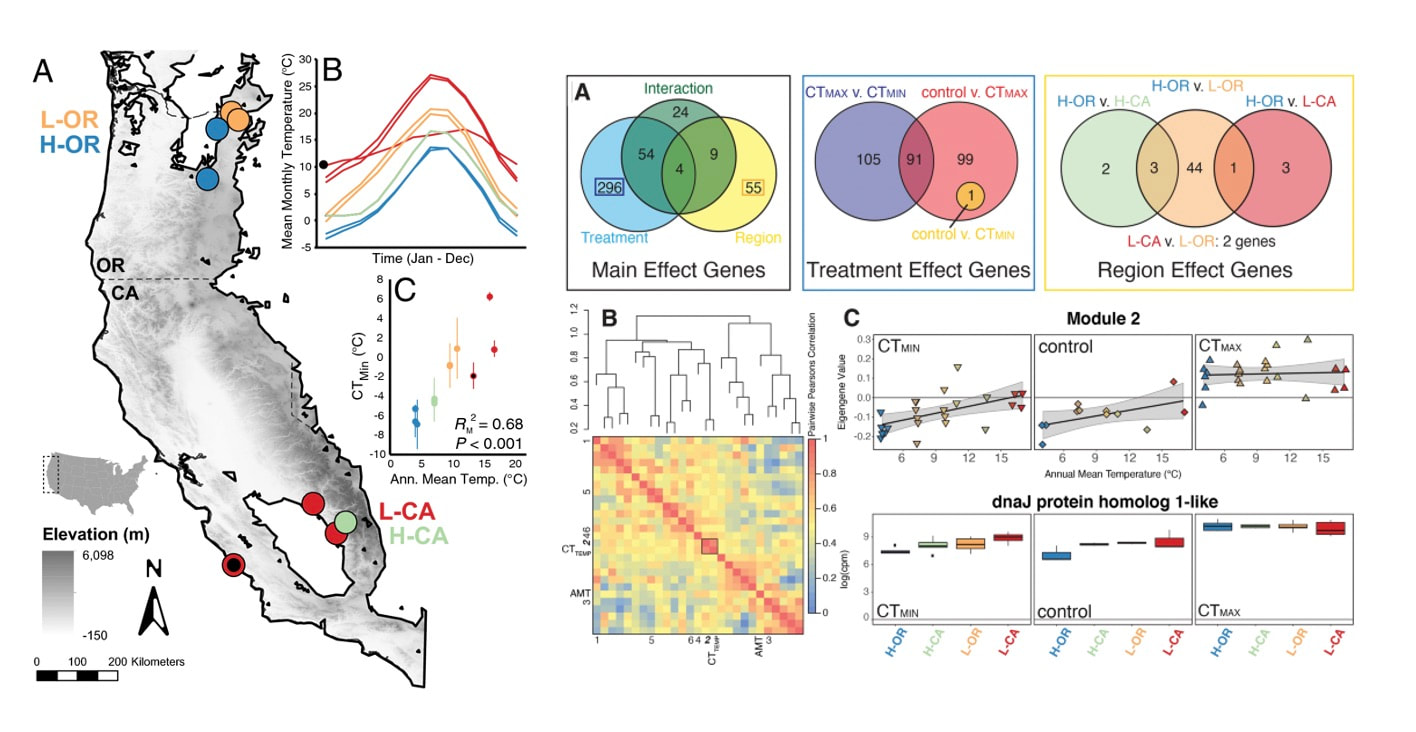 Pimsler, M.L., Oyen, K.J., Herndon, J.D. et al. Biogeographic parallels in thermal tolerance and gene expression variation under temperature stress in a widespread bumble bee. Sci Rep 10, 17063 (2020). https://doi.org/10.1038/s41598-020-73391-8
|
Lozier Lab NewsDispatches from the lab and field! Archives
March 2023
Categories
All
|




 RSS Feed
RSS Feed